Amplicon pileup analysis pipeline
[1/4] Setup environment
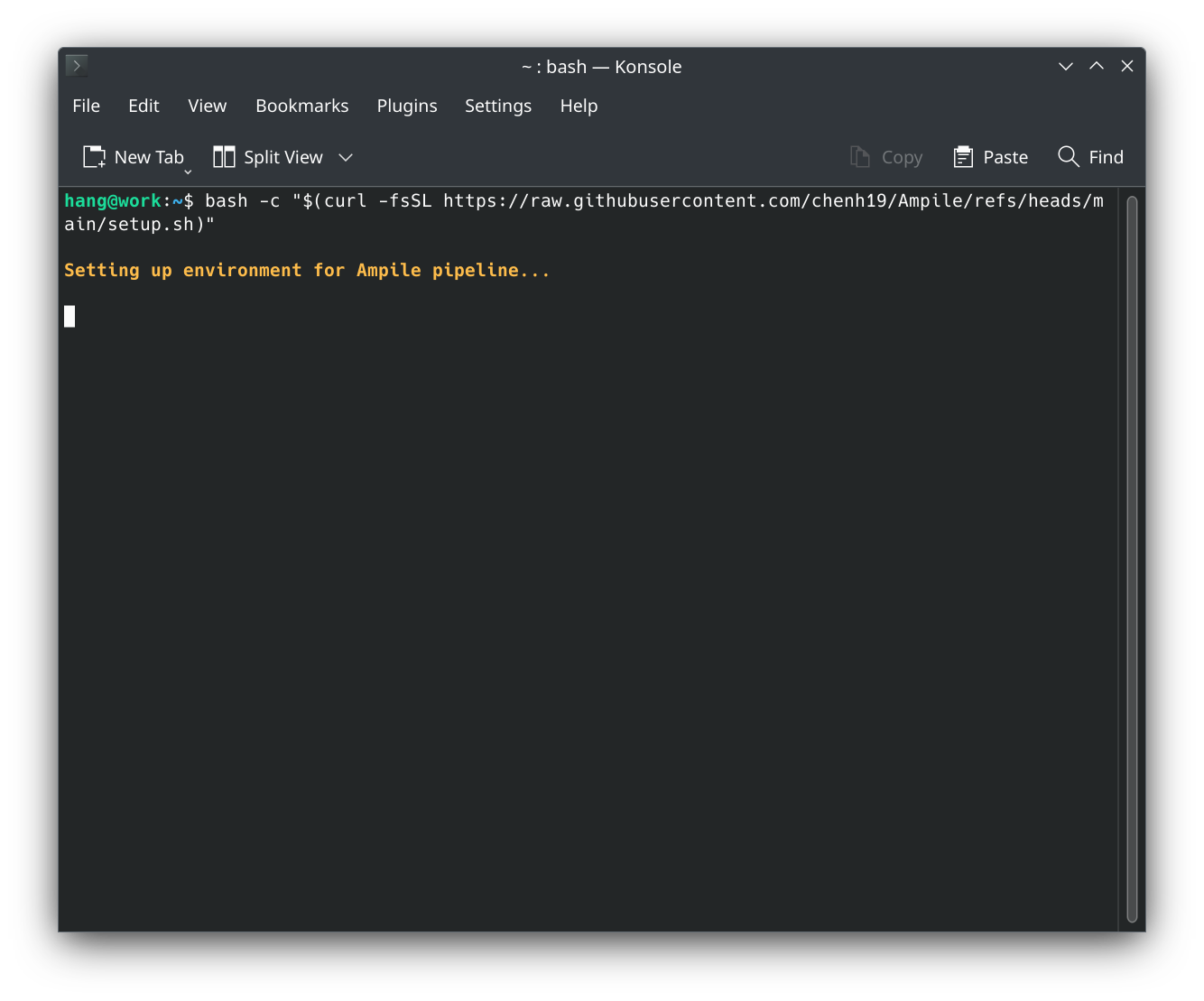
Pipeline failures are often due to an improperly configured environment. To ensure a robust and consistent setup for Ampile, I’ve created a dedicated configuration script. To execute the setup:
- Install
curlif it is not already installed on your system (e.g.,sudo apt install curlon Ubuntu). - Connect to internet and execute the below command in terminal:
bash -c "$(curl -fsSL https://raw.githubusercontent.com/chenh19/Ampile/refs/heads/main/setup.sh)"
Note: (click to expand)
- This pipeline is dependent on:
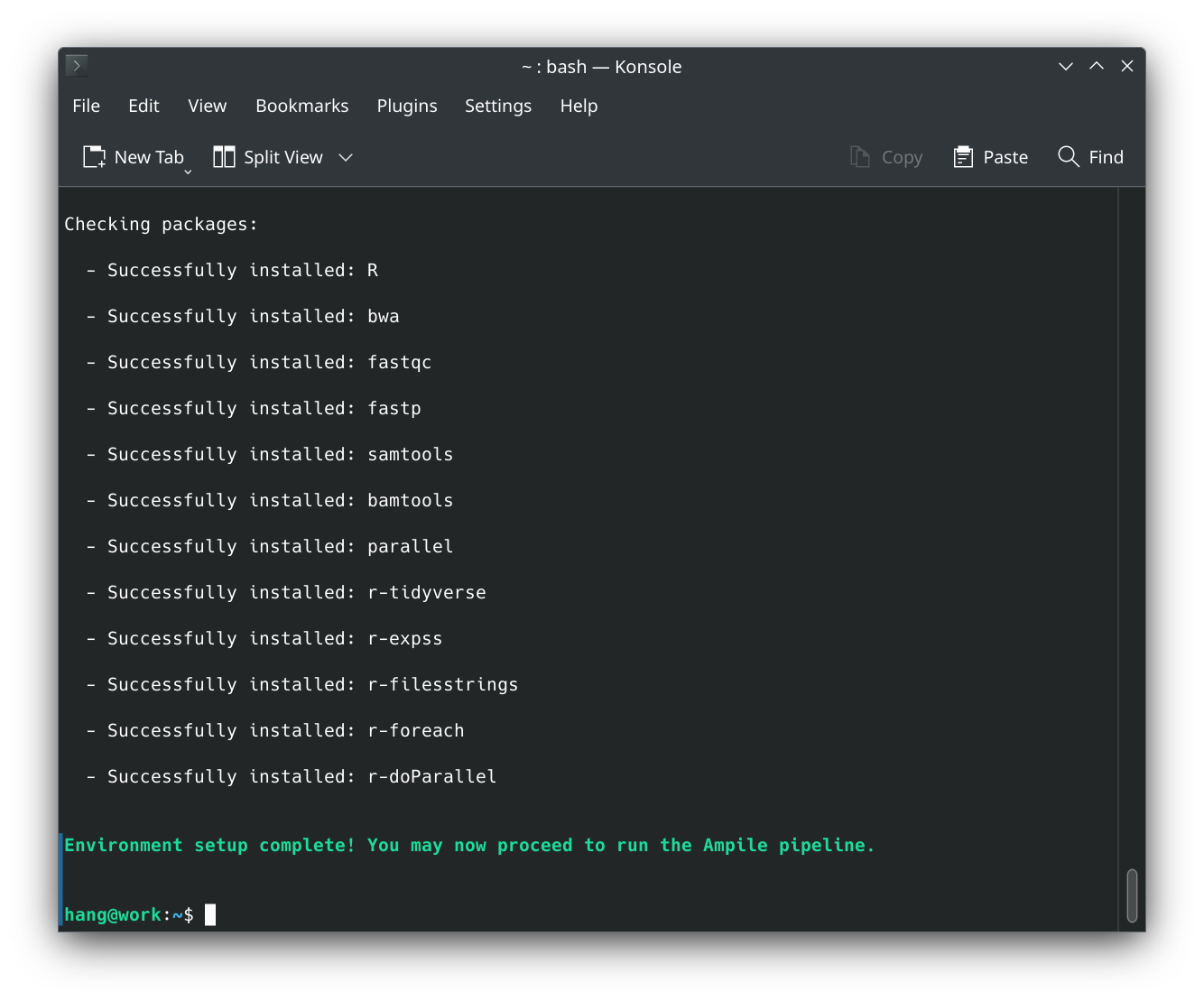
R,bwa,fastqc,fastp,samtools,bamtools,parallel,r-tidyverse,r-expss,r-filesstrings,r-foreach,r-doParallel. It can be run in Linux, FreeBSD, and MacOS environments. - The ampile.sh script will verify that all required packages are installed before proceeding with the analysis.
- The setup.sh script requires no directory changes, and does not need administrative privileges on Linux.
- The pipeline has been tested on Debian 12, Ubuntu 24.04, Kubuntu 24.04, KDE Neon 20250616, Linux Mint 22.1, Zorin OS 17.3, Pop!_OS 22.04, Elementary OS 8, Fedora 42, Rocky Linux 10, AlmaLinux 9, RHEL 8 (UChicago Midway3), CentOS 7 (UChicago Midway2), FreeBSD 14.3, and MacOS Sequoia. If you’re using an unsupported OS or prefer an alternative setup method, please ensure that all required dependencies are installed.
[2/4] Prepare input files
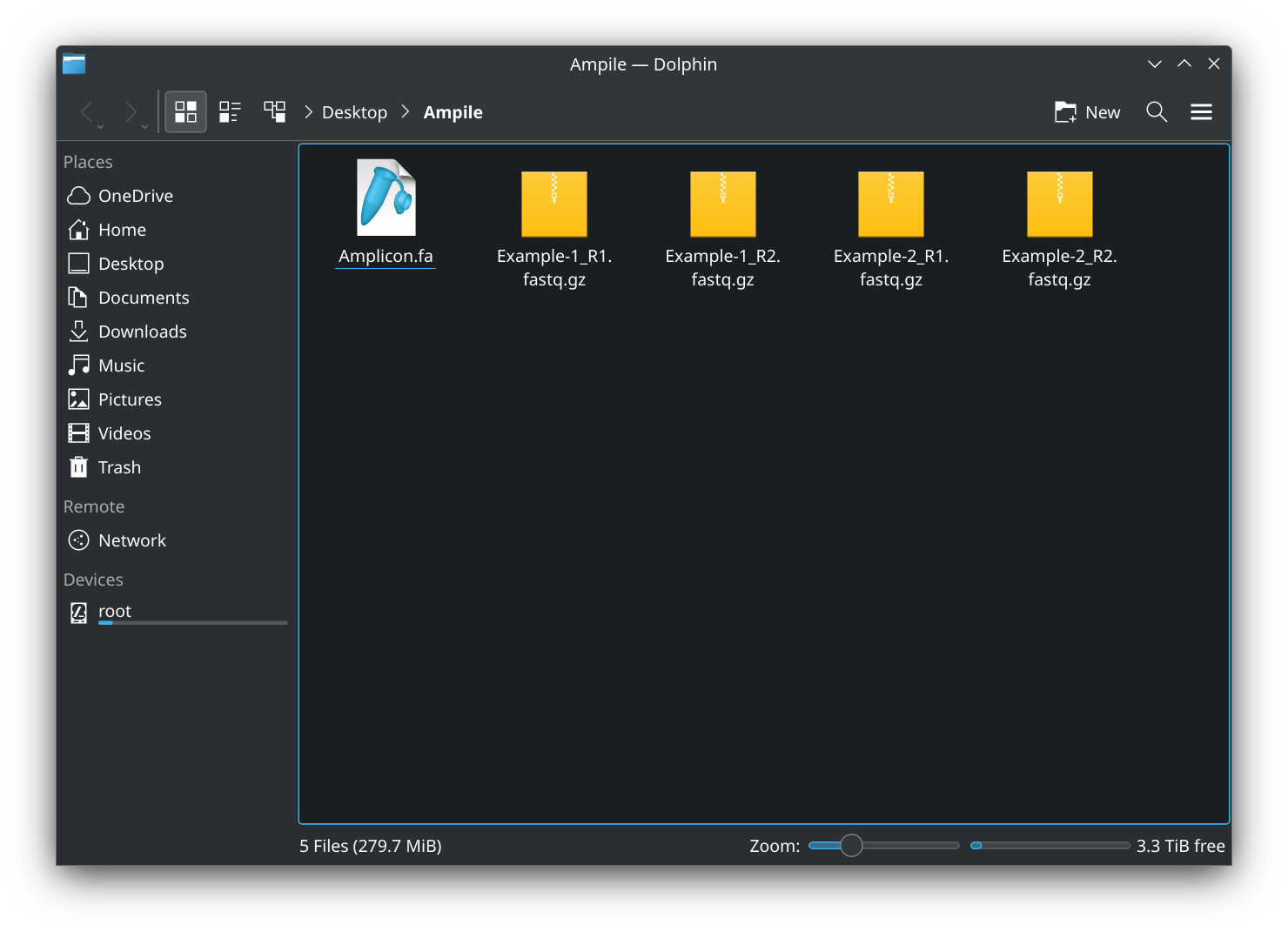
- Prepare reference sequences (
.fafiles) and sequencing reads (.fastqor.fastq.gzfiles) in a master folder (you may name the folder as desired):
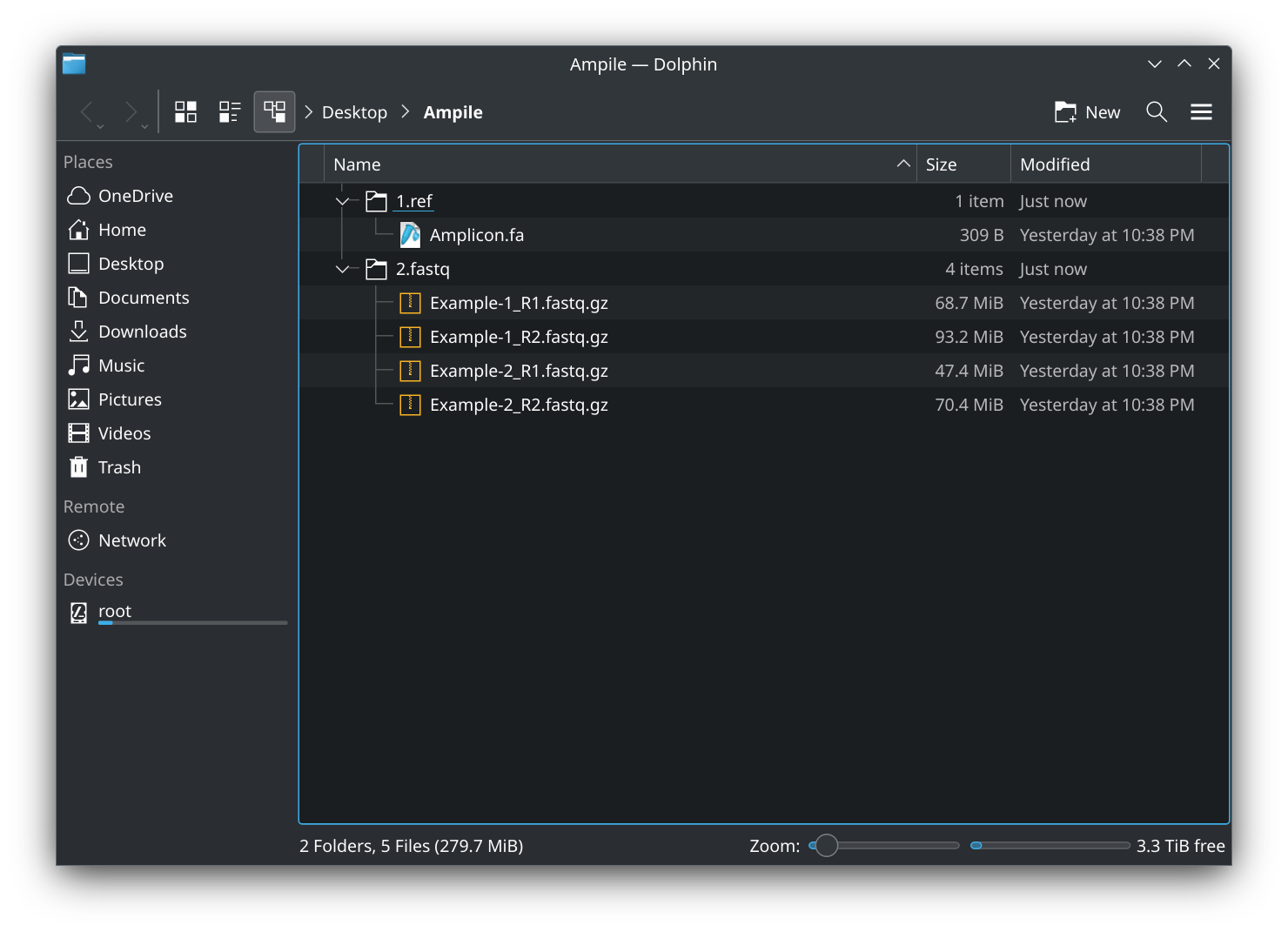
- You may also organize the files into the two designated subfolders,
./1.ref/and./2.fastq/:
Note: (click to expand)
- Example files are provided in the /examples/ folder.
- The pipeline will automatically organize input files if they are not already in the two designated subfolders.
- The pipeline will also automatically compress sequencing reads to
.fastq.gzif they are provided in.fastqformat.
[3/4] Running the pipeline
- Change current directory to the folder containing the input files (e.g.,
cd ~/Desktop/Ampile/). - Connect to internet and execute the below command in terminal:
bash -c "$(curl -fsSL https://raw.githubusercontent.com/chenh19/Ampile/refs/heads/main/ampile.sh)"
- Alternatively, you may download the GitHub repository and place all scripts in the /src/ folder along with the input files to run them manually:
Note: (click to expand)
- All scripts assume the master folder as the working directory.
- If you are running the scripts manually on Linux, please don’t forget to load conda environment first:
source ~/miniconda3/etc/profile.d/conda.sh && conda activate ampile
[4/4] Done
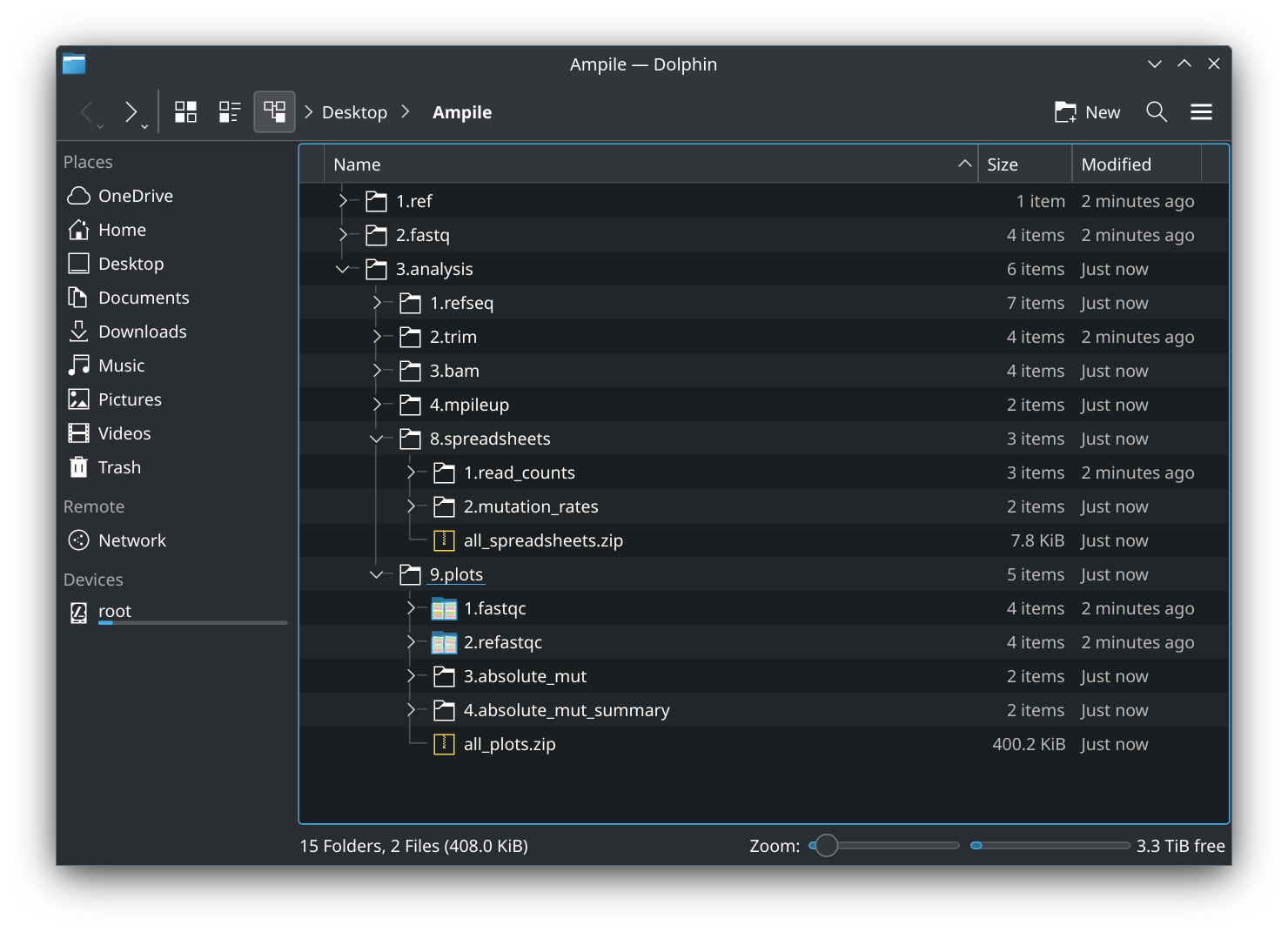
- You may further analyze the parsed mutation rates and perform comparative analyses between groups. The corresponding spreadsheets are located at
./3.analysis/8.spreadsheets/2.mutation_rates/.
Note: (click to expand)
- The directories
./3.analysis/1.refseq/,./3.analysis/2.trim/,./3.analysis/3.bam/, and./3.analysis/4.mpileup/contain large intermediate files. You may choose to delete them unless you need them for troubleshooting.